Lipid nanoparticles (LNPs) are in vivo leading systems delivered for therapeutic applications as small interfering RNAs (siRNAs). The formulation of LNP siRNA systems requires rapid mixing of solutions containing cationic lipids with solutions containing siRNA. Current formulation procedures employ macroscopic mixing processes to produce systems with a diameter of 70 nm or larger with variable siRNA encapsulation efficiency, homogeneity, and reproducibility. Here, we show a microfluidic mixing technique that allows for millisecond mixing at nanoscale, reproducibly generating LNP siRNA systems with a limit size of 20 nm or larger and substantially complete encapsulation of siRNAs under a wide range of conditions with a polymorphism index as low as 0.02. The optimized LNP siRNA system generated by microflow mixing achieved 50% target gene silencing in mouse hepatocytes at a dose level of 10 μg/kg siRNA.
Lipid nanoparticles (LNPs) are the primary system used in therapeutic applications for the delivery of small interfering RNAs (siRNAs) in vivo in vivo applications. LNP siRNA systems can be used in a variety of animal models to treat therapeutic genes by intravenous silencing and are being used in clinical trials for the treatment of cardiovascular disease, liver cancer, and other diseases. Several methods have been developed to manufacture LNP siRNAs, including mixing pre-formed vesicles (PFVs) with siRNA or using a T-tube mixer to mix lipids dissolved in ethanol with siRNA aqueous solutions. These methods produce LNPs with a diameter of 70 nm or larger with siRNA encapsulation efficiency of 65-95%. However, because macroscopic mixing methods can lead to uneven local mixing rates, they tend to produce LNPs with high polydispersity and poor batch-to-lot repeatability. Microfluidic mixing allows for rapid mixing at the millisecond and nanoliter levels, and in this article we demonstrate the use of microfluidic mixing to reliably prepare previously unattainable LNP siRNA systems. We employ a simple but efficient microfluidic device, the Staggered Herringbone Micromixer (SHM), which has a higher yield relative to other micromixer geometries, making it suitable for large-scale production of LNP-siRNA.
Microfluidic mixing results in the generation of monodispersive LNPs. Structured chaotic advection (SHM) provides a reproducible and very rapid mixing of two input streams under moderate Reynolds number conditions (2 < Re < 500). After combining the two fluid flows, they induce a rotational flow through a series of herringbone structures so that the fluids wrap around each other and the direction changes between half a cycle. This results in a chaotic flow signature characterized by exponentially reducing the characteristic diffusion length between the two liquids and enabling rapid advection mixing. When the total flow rate is 2 ml/min, the SHM employed in this paper can achieve complete mixing in 3 milliseconds.
To develop a manufacturing process for LNP siRNAs, we employed a test formulation based on previous studies, in which the LNP-siRNA system was prepared using a PFV and T-tube hybrid process. The lipid composition includes ionizable cationic lipids (DLinKC2-DMA), 1,2-distearayl-sn-glyceryl-3-phosphocholine (DSPC), cholesterol, and PEG-lipid, with cationic lipids ranging from 40 to 60 mol% and PEG-lipid content ranging from 1 to 5 mol%. The siRNA/total lipid ratio was maintained at 0.06 (wt/wt). DLinKC2-DMA has a significant pKa value of 6.76, is able to form complexes with siRNA at low pH (e.g., pH 4.0), and makes the LNP surface charge nearly neutral at physiological pH, reducing toxic side effects. The test formulation consisted of DLinKC2-DMA/DSPC/cholesterol/PEG-c-DMA in a molar ratio of 40:11.5:47.5:1 with a siRNA/lipid ratio of 0.06 (wt/wt). The PEG-lipid content, cationic lipid content, and/or siRNA/lipid ratio were subsequently changed to optimize LNP size or potency.
We first investigated whether self-assembled mixing (SHM) can produce a monodisperse LNP siRNA system with a definite size at a range of solute concentrations. We hypothesize that at a sufficiently fast mixing rate, lipid nanoparticles (LNPs) formed by precipitation due to a rapid rise in the polarity of the medium will adopt a Z-small or “limit size” compatible with the physical properties of the component’s molecules. Therefore, at faster flow rates, a progressively shorter mixing time is expected to result in increasingly monodisperse limit-size LNP siRNA systems. We generated the LNP siRNA system from the test formulation with a total flow rate of 0.02 to 4 ml/min. Measured by dynamic light scattering, the LNP diameter remains constant at flow rates above 0.2 ml/min (digital-weighted mode; reflecting the average diameter derived from the apparent LNP molecular weight). However, at a high total flow rate of 4 ml/min at Z, a highly monodispersive LNP siRNA system with a polydispersity index (PDI) of 0.03 or less was achieved for terminal siRNA concentrations ranging from 0.25 mg/ml to 0.59 mg/ml.
Parallelization of microfluidic mixers allows for scaling of LNP manufacturing A viable LNP siRNA formulation process must also be scalable. Scaling can be challenging due to mass transport differences as the total volume of the reaction increases. The inherent advantage of microfluidic mixing is that it can be easily scaled with parallelized mixing equipment (see Figure 1b). To demonstrate this, we used a setup with six parallel SHM elements to produce limit-size LNPs consisting of 1-palmitoyl-2-oleoyl PC (POPC)/cholesterol at 72 mL/min or 580 mg LNP/min (see Supplementary Figures S3 and S4). To our knowledge, this is the Z-rate LNP synthesis demonstrated so far using microfluidic methods and performs well compared to alternative scale-up processes.
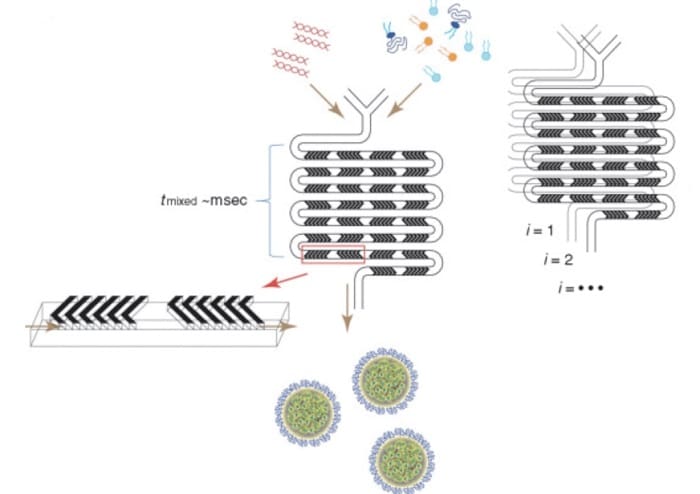
Figure 1. Schematic diagram of lipid nanoparticle (LNP) small interfering RNA (siRNA) formulation strategy using a staggered herringbone micromixer (SHM). (a) Lipids in ethanol and siRNA in aqueous solution are pumped into the two inlets of the microfluidic mixing device through syringe pumps. The herringbone structure induces chaotic advection of laminar flow, resulting in rapid mixing of ethanol and aqueous phases, and correspondingly rapidly increasing the polarity of the lipid solution. LNPs are formed by precipitation at critical polarity. (b) Parallelization of microfluidic mixers to achieve formulation scale-up while maintaining the same production conditions. This is achieved through vertical (i = 1, 2,..) and horizontal (j = 1, 2,…) mixer replication, fluid handling through pipes on the chip. The hybrid channels are 200 μm × 79 μm in size and 31 μm high and 50 μm thick in herringbone structure.
It is important that LNP siRNA systems generated by microfluidic mixing exhibit strong gene silencing in vivo. We need to demonstrate that these LNP siRNA systems generated by microfluidic mixing have the same or higher potency than previous systems generated. To date, the reported Z-effective siRNA preparations are LNP systems prepared by T tube synthesis using DLinKC2-DMA cationic lipids and optimized cationic lipid ratios and siRNA/lipid ratios. It has been confirmed that these LNP siRNA systems can achieve 50% gene silencing in mouse FVII models with dose levels (ED50) as low as 0.02 mg siRNA/kg body weight. Therefore, the gene silencing potency of the LNP FVII-siRNA system generated by microfluidic mixing at different cationic lipid masses was first measured in a mouse FVII model, and then as a siRNA/lipid ratio function.
The results of this study suggest that the presence of SHM in a microfluidic mixing device can be used to generate LNP systems with predictable “limit” sizes. siRNA can be efficiently encapsulated in these systems, and the resulting LNP siRNA systems exhibit excellent gene silencing in vivo. We distinguish the results presented here from previous LNP systems using microfluidic mixing techniques, then discuss the mechanism of LNP and LNP siRNA systems using microfluidic mixing devices, and finally point out the advantages of using SHM geometries for microfluidic mixing compared to previous macroscopic mixing procedures for LNP siRNA systems.
Considerable work has been done to generate lipid and polymer nanoparticles, or lipid complexes containing plasmid DNA, using microfluidic mixing, mainly through hydrodynamic focusing geometry. While the results are encouraging, there are some limitations, such as limitations in the size and quantity of materials produced. For example, a 50 nm diameter limit size system under hydrodynamic focus is only possible at a flow rate ratio of 30 or higher, in which case it results in a large degree of material dilution. The results we provide demonstrate that SHM can produce high amounts (500 mg/min) of LNP siRNA systems with a limit size range of 20-100 nm, and their limit size can be controlled by simply changing the PEG-lipid content.
It is worth noting that as we completed the preparation phase of this paper, Chen et al. published an article stating that they also utilized microfluidic mixing techniques and SHM microstirrers to produce various types of LNP siRNA formulations on a small scale to determine suitable for vivo delivery of new components. This work employed low flow rates (~0.3ml/min), low encapsulation efficiency (~80%), and LNP sizes of 70-80nm.
We are interested in the mechanism by which the miniaturization process allows the formation of LNPs and siRNAs containing LNPs: first, how to form LNPCs of 100 nm or smaller; secondly, how to make siRNAs close to 100% encapsulation efficiency. Regarding LNPs formation, the mixing rate is clearly an important parameter. The rapid mixing of ethanol-lipid solution with water buffer leads to a rapid increase in the polarity of the medium, resulting in a high supersaturation state across the entire level, resulting in rapid and homogeneous nucleation of nanoparticles. These nucleation events are very rapid to capture unstable sublimit size particles whose time scale affects particle morphology/structure, and then merge into branch-bound size particles, which are determined by the spatial constraints provided by the component. Appropriate lipid components and their relative quantities are selected, and then controlled diffusion into further growth.
There are two points of interest regarding the mechanisms by which microfluidic processes allow LNPs and LNP-containing siRNAs to form: firstly, the mechanism by which LNPs of size are 100 nm or smaller, and secondly, by which siRNAs can be encapsulated with near-100% efficiency. Regarding LNP formation, the mixing rate is clearly an important parameter (as shown in this and other studies). The rapid mixing of the ethanol-lipid solution with the water buffer results in a rapid increase in the polarity of the medium, highly supersaturating the lipid monomers throughout the mixing volume, resulting in rapid and uniform nanoparticles. These nucleation events are much faster than the particle formation/aggregation timescale and result in the formation of non-thermodynamically stable submarginal size particles. These submarginal size particles then condense to form defined size particles, which are determined by the steric resistance and energy constraints applied by the providing components. Appropriate selection of lipid components and their relative numbers can control the size of the Z-terminal LNP (as shown in this paper by changing the amount of PEG-lipid; PEG-lipid preferentially exists outside the LNP and confers stability, and inhibits subsequent growth through further lipid monomer binding).
It was observed that the LNP siRNA system prepared with microfluidic hybrid technology exhibited nearly 100% siRNA encapsulation efficiency, indicating that as the polarity of the medium increased, the cationic lipids precipitated to initially react with the siRNA nucleic acid, and then were encapsulated by PEG-lipid when the polarity was further increased. As previously described, this model is consistent with the observation of low-temperature transmission electron microscopy that the LNP siRNA system presents a “solid-state core” electron-dense appearance, which differs from the bilayer vesicle system in that it has a circular structure and a lower internal electron density. The molecular modeling method and the physical characteristics such as the density exhibited by these LNP siRNA systems also support the existence of nanostructured lipid digital cores.
It is important to note that in-line mixing techniques like SHM mixers require lipids to be soluble in the organic solvents used. Lipids that are less soluble in ethanol may need to heat the solvent to achieve sufficient solubility, or other water-compatible organic solvents can be employed. Microfluidic mixers such as SHM can be constructed from a variety of materials that are resistant to most solvents.
The microfluidic formulation process offers several advantages over previous LNP siRNA synthesis using macroscopic mixing techniques such as PFV method and T-tube hybrid technology. The preparation of LNP siRNA systems through microfluidic processes allows for higher encapsulation efficiency, production of smaller LNP systems, and the ability to be produced in small batches with little loss (device dead volume of approximately 1 μl). In addition, PFVs do not need to be produced.
Microfluidic methods also include the following advantages over T-tube mixers: the ability to produce smaller models of systems with diameters of less than 50 nm (T-tube methods report a small Z diameter of 50 nm) and the use of high flow rates of less than 1 ml/sec for rapid mixing is not necessary. Microstirrers, on the other hand, allow for LNP siRNA formulation at lower flow rates under well-defined, reproducible conditions, with minimal loss due to the low dead volume and simple preparation of small batches for LNP optimization and ex vivo testing.
The move from benchtop experimental preparation to large-scale LNP siRNA manufacturing is also a major advantage by parallelizing microfluidic stirring equipment.
In conclusion, the results presented here show that microfluidic stirring using SHM microstirrers enables the routine production of LNP siRNA systems in the 20-100 nm range, and offers many advantages such as ease of design, oligomeric dispersion, high siRNA encapsulation efficiency, improved scalability, and comparable or even better gene silencing potency than previous processes. The ability to form LNPs with a diameter of 50 nm or less is important because such systems exhibit enhanced penetration of target tissues (e.g., tumors); at the same time, the formation of LNP systems at a low proportion of more than three times significantly facilitates the production of LNPs at ideal scale. It is expected that SHM microfluidic mixing will become the preferred LNP synthesis technique in laboratory and clinical settings.
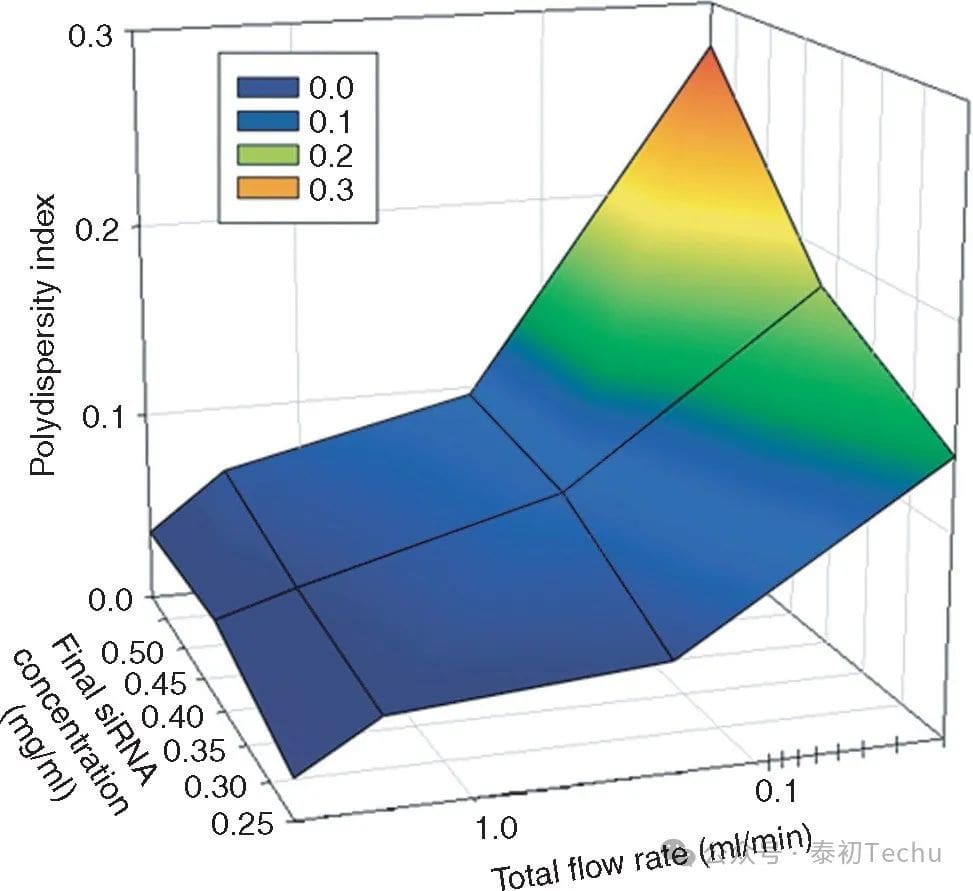
Figure 2. The high total flow rate of the staggered herringbone micromixer (SHM) reduces the polydispersity of lipid nanoparticles (LNPs) small interfering RNAs (siRNAs). The polydispersity index (PDI) is dependent on the total flow rate and the concentration of lipids and siRNAs in the ethanol and aqueous phases, respectively. PDI is determined by the second-order coefficients in the cumulative analysis provided by dynamic light scattering (DLS) (PDI = (σ/μ)2). The total flow rate ranged from 0.02 to 4 ml/min, keeping the water buffer-to-ethanol volume-flow ratio constant at 3:1. The aqueous siRNA concentration ranged from 0.25 to 0.59 mg/ml, while the lipid concentration ranged from ~4 to 10 mg/ml, keeping the siRNA/total lipid ratio constant at 0.06 wt/wt. The PDI value represents the average of the 4 measurements. The lipid components used were DLinKC2-DMA/DSPC/CHOLESTEROL/PEG-C-DMA, with a molar ratio of 40:11.5:47.5:1.water buffer of 25 mmol/l acetate, pH 4.DSPC, and 1,2-distearayl-sn-glycerol-3-phosphocholine.
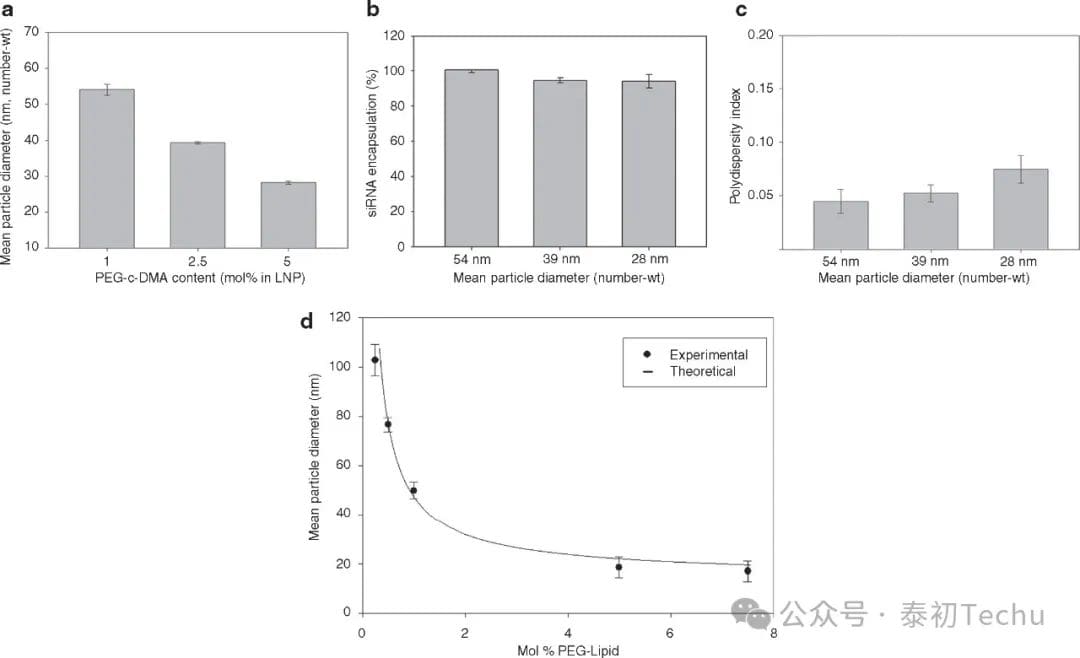
Figure 3. Increasing PEG-c-DMA content produces progressively smaller lipid nanoparticles (LNPs) of small interfering RNA (siRNA) systems. (a) Effect of PEG-c-DMA content on LNP size under fast mixing conditions (4 ml/min total flow rate, siRNA buffer:lipid-ethanol volume-to-flow ratio of 3:1). LNP consists of DLinKC2-DMA/DSPC/cholesterol/PEG-c-DMA with molar ratios of 40:11.5:47.5:1, 40:11.5:46:2.5, and 40:11.5:43.5:5, and PEG-c-DMA of 1,2.5, and 5 mol%, respectively. The siRNA-total lipid ratio of LNP is 0.06 wt/wt. (b) Encapsulation efficiency by increasing the PEG-c-DMA content from 1 mol% to 5 mol% as the LNP size decreased from 42 nm to 26 nm. The LNP sample was dialyzed in phosphate buffer (PBS) before measuring the encapsulation. Encapsulation refers to the percentage of siRNA present in the LNP after removal of the free siRNA using an anion-exchange spin column. (c) Polydispersity of the LNP by increasing the PEG-c-DMA content from 1 mol% to 5 mol% as the LNP size decreases from 54 nm to 28 nm. The polydispersity index (PDI) is determined as shown in the Figure 2 legend. (d) The size of the empty LNP as a function of the PEG lipid content ranges from 0.25-5 mol%. The LNP consists of DLinKC2-DMA/DSPC/cholesterol/PEG-c-DMA, which is maintained at 40 and 11.5 mol% for DLinKC2-DMA and DSPC, respectively. The titration of PEG-c-DMA is compensated by adjusting for cholesterol. All LNPs were produced in the lipid-ethanol phase at an initial lipid concentration of 20 mmol/l and then mixed with 25 mmol/l acetic acid buffer at pH 4. The numerically weighted average diameter shows the average diameter of the LNPs after dialysis of PBS to remove residual ethanol and increase the pH to 7.4. The error column represents the standard deviation of the mean (n = 3). DSPC, 1,2-distearacyl-sn-glycerol-3-phosphocholine.
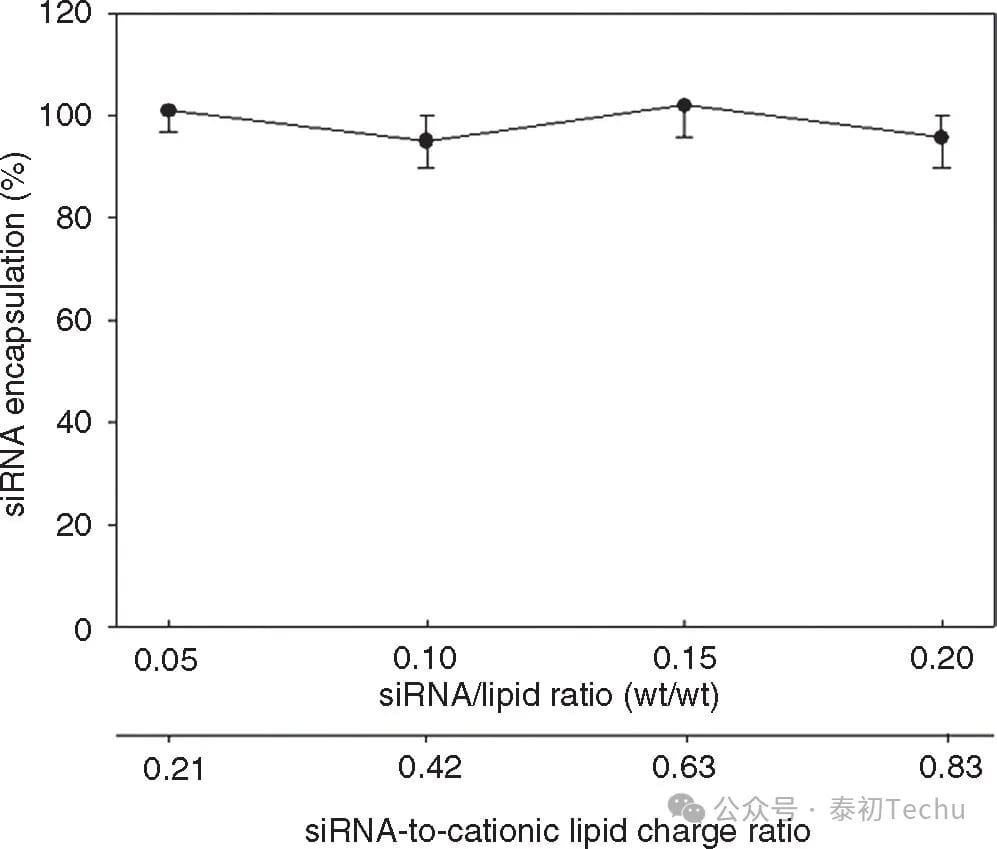
Figure 4. Lipid nanoparticles (LNPs) The formulation of small interfering RNA (siRNA) uses microfluidic mixing to efficiently encapsulate over a wide range of siRNA-cationic charge ratios. The LNP consists of Dlin-KC2-DMA/DSPC/CHOLESTEROL/PEG-C-DMA with a molar ratio of 40:11.5:47.5:1 and a siRNA-to-total lipid ratio of 0.06 wt/wt. The total flow rate was maintained at 2 ml/min using 10 mmol/l lipid-ethanol phase mixed with water buffer containing siRNA (25 mmol/l acetate, pH 4). Encapsulation refers to the percentage of siRNA in the LNP after removal of free siRNA using an anion-exchanged spin column. Error bars represent the standard deviation of encapsulation measured from the three LNP formulations. DSPC, 1,2-distearacyl-sn-glyceryl-3-phosphocholine.
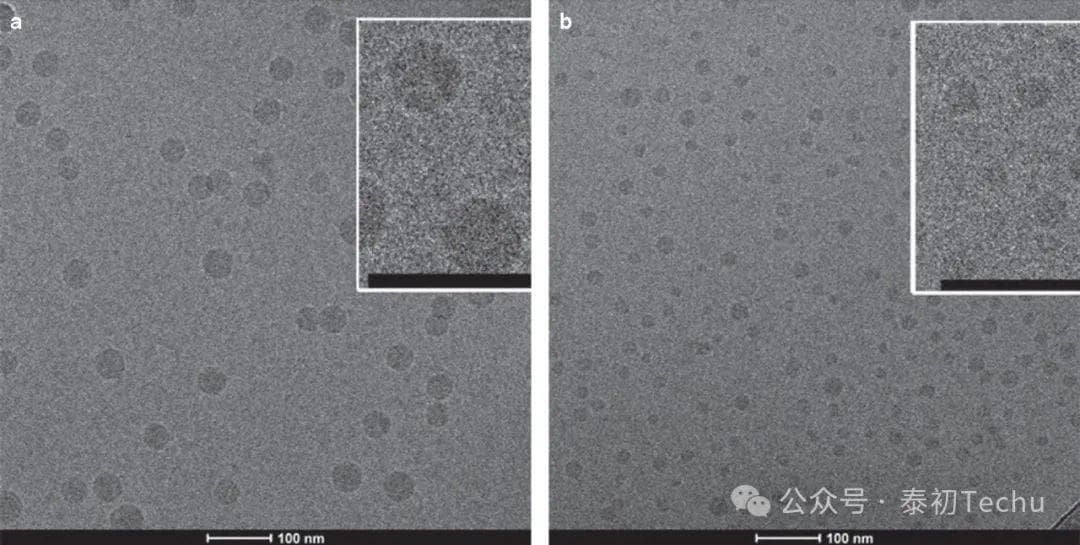
Figure 5. Cryo-transmission electron microscopy (cryo-TEM) of a lipid nanoparticle (LNP) small interfering RNA (siRNA) system containing 1 and 5 mol% PEG-c-DMA. (a) Cryo-TEM micrograph (wt/wt) of LNP siRNA composed of DLinKC2-DMA/DSPC/cholesterol/PEG-c-DMA (molar ratio 40:11.5:47.5:1) and siRNA. (b) cryo-TEM micrograph (wt/wt) of LNP siRNA composed of DLinKC2-DMA/DSPC/CHOLESTEROL/PEG-C-DMA (MOLAR RATIO 40:11.5:43.5:5) and siRNA. LNP imaged at 50K magnification. The LNP formulation was performed under fast mixing conditions (4 ml/min total flow, siRNA buffer:lipid-ethanol volume-to-flow ratio of 3:1) with a staggered herringbone microstirrer (SHM) with the ethanol phase containing 30 mmol/l lipid. The LNP siRNA dispersion was concentrated prior to imaging. The scale bar represents 100 nm. DSPC, 1,2-distearacyl-sn-glyceryl-3-phosphocholine.
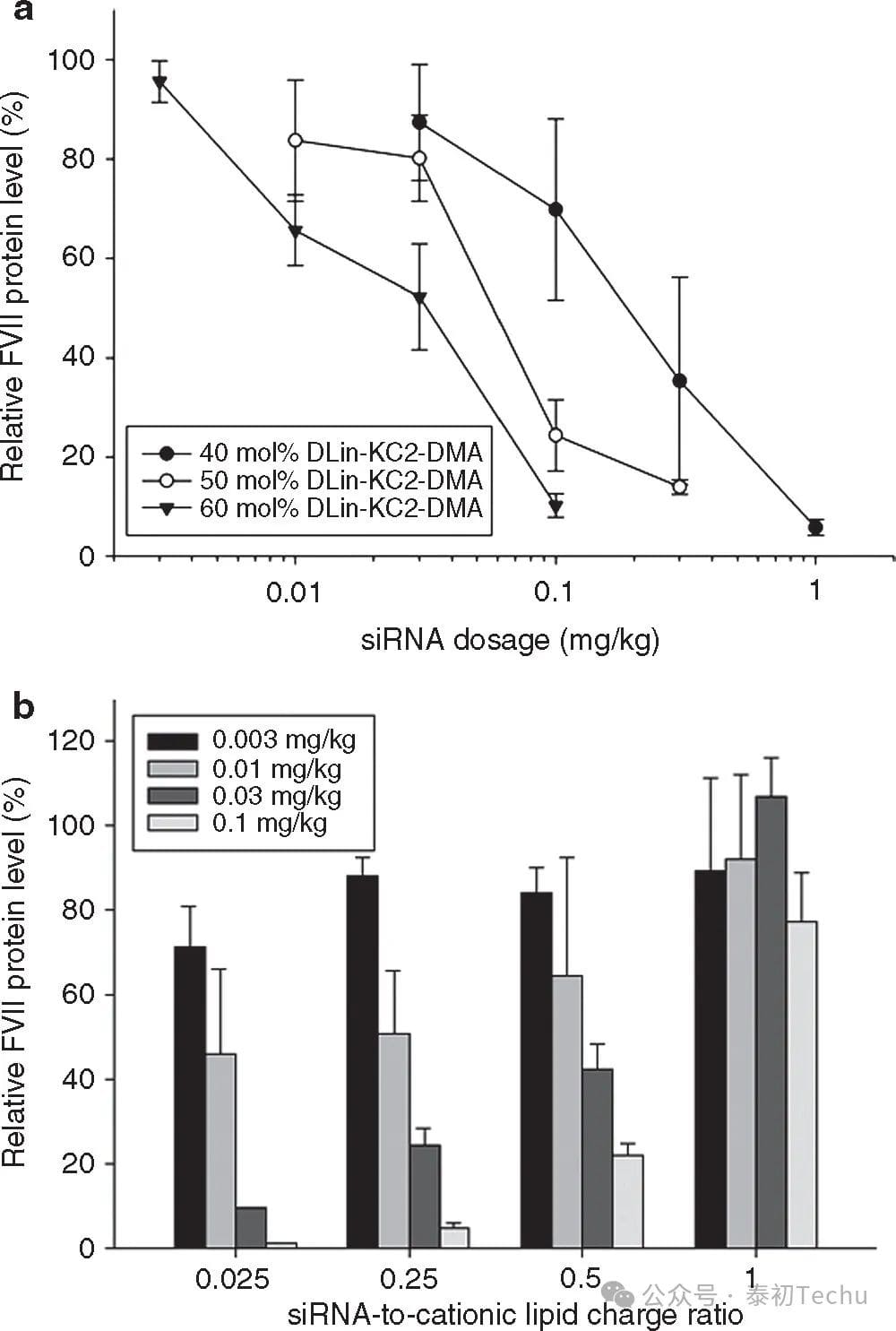
Figure 6. Optimization of the silencing efficacy of lipid nanoparticles (LNPs) small interfering RNA (siRNA) genes as a function of cationic lipid content and siRNA/total lipid ratio in an FVII mouse model. (a) Effect of LNP DLinKC2-DMA content on FVII gene silencing in an FVII mouse model. FVII expression was monitored after intravenous injection of an LNP siRNA system containing 40 to 60 mol% DLinKC2-DMA for 24 h. PEG-c-DMA content was maintained at 1 mol%, and the addition of cationic lipids was compensated by reducing DSPC and cholesterol content, keeping the DSPC-to-cholesterol ratio at 0.25 (mol/mol). (b) Effect of siRNA/total lipid ratio changes on FVII gene silencing in FVII mouse models. The lipid components used were DLinKC2-DMA/DSPC/CHOLESTEROL/PEG-C-DMA (MOLAR RATIO 60:7.5:31.5:1). The siRNA/total lipid ratio varied from 0.01 to 0.35 (wt/wt), corresponding to siRNA-to-cationic lipid charge ratios of 0.025, 0.25, 0.5, and 1, respectively. LNP siRNA was systematically administered to mice via tail vein injection (n = 3/dose level). Blood was collected 24 h after injection and factor VII levels were determined by colorimetry.
References:
Molecular Therapy – Nucleic Acids ( IF 8.8 ) Pub Date : 2016-12-14 , DOI: 10.1038/mtna.2012.28